新闻 – 第 10 页 – 卫材(中国)药业有限公司-威尼斯人888
卫材和渤健今日宣布,在加州旧金山举行的2022年阿尔茨海默病临床试验(ctad)会议上,卫材将公布仑卡奈单抗(开发代码:ban2401)大型全球iii期验证性clarity ad临床研究的结果,试验药物仑卡奈单抗是治疗脑内确认存在淀粉样蛋白病理的阿尔茨海默病(ad)所致轻度认知障碍(mci)和轻度ad(统称为早期ad)的抗淀粉样蛋白 (aβ) 原纤维抗体。
ctad关于仑卡奈单抗的大会演讲摘要
clarity ad研究设计
卫材clarity ad是一项全球验证性iii期安慰剂对照、双盲、平行组、随机试验,在北美、欧洲和亚洲的235个研究中心纳入了1,795例早期ad患者(仑卡奈单抗组:898例;安慰剂组:897例)。按1∶1将患者随机分组,两组分别接受每两周一次的10 mg/kg静脉给药的安慰剂或仑卡奈单抗治疗,并根据临床亚组(ad或轻度ad引起的mci)、基线时是否给予已获批的ad对症治疗药物(如乙酰胆碱酯酶抑制剂、美金刚或联用这两种药物)、apoe4携带状态和地理区域对随机化进行分层分析。患者纳入标准包括但不限于高血压、糖尿病、心脏病、肥胖、肾脏疾病和抗凝药物治疗。由于卫材在clarity ad研究中采用了多元化的招募策略,美国随机分组的患者中分别有4.5%和22.5%的非裔与西班牙裔。
研究主要终点是临床痴呆综合评定量表1 (cdr-sb,clinical dementia rating sum of boxes)和整体认知与功能量表评分较基线的变化值,关键次要终点是淀粉样正电子发射断层扫描(pet) centiloids、ad评估量表-认知功能子表14 (adas-cog142)、ad综合评分(adcoms3)和阿尔兹海默病协作研究组 mci 日常活动表(adcs mci-adl4)较基线的变化值。此外,在可选亚组研究中评估了通过tau pet (n=257)和ad病理的脑脊液(csf)生物标志物(n=281)测量的脑tau病理学的纵向变化。
clarity ad有效性结果
仑卡奈单抗组和安慰剂组治疗18个月后,主要终点cdr-sb较基线的平均变化分别为1.21和1.66分。仑卡奈单抗治疗18个月较安慰剂显著改降低体认知与功能量表评分0.45分(95%置信区间(ci):-0.67~ -0.23; p=0.00005),降幅达27%。从治疗6个月开始的所有时间点,治疗组较安慰剂组的绝对差每3个月增加1次,治疗组的cdr-sb较基线有高度统计学显著差异(所有p值均<0.01)(图1)。
相比安慰剂组,治疗组所有关键次要终点也显示出高度统计学显著差异(p<0.001=。在淀粉样pet亚组试验中,仑卡奈单抗治疗组自治疗3个月开始就显著减轻淀粉样斑块负担。治疗组和安慰剂组的centiloids平均变化值分别为-55.5和3.6(平均差: -59.1 [95%ci:-62.6, -55.6]; p<0.00001)。基于adas-cog14评分,仑卡奈单抗治疗18个月后延缓了26%的认知功能减退(平均差: -1.44 [95%ci: -2.27, -0.61]; p=0.00065)。基于adccoms评估,仑卡奈单抗治疗18个月后延缓了24%的疾病进展(平均差: -0.050 [95% ci: -0.074,-0.027; p=0.00002])。基于adcs mci-adl评分,仑卡奈单抗治疗18个月后延缓了37%的日常生活能力减退(平均差: 2.016 [95%ci: 1.208, 2.823]; p<0.00001)。此外,主要分层分析显示,在所有疾病分期亚组中(ad所致mci或轻度ad)、apoe4携带(未携带或携带)、是否联用已获批的ad对症治疗药物以及所属地理区域(北美洲、亚洲或欧洲),仑卡奈单抗治疗18个月的cdr-sb、adas-cog14和adcs mci-adl改善结果均一致。
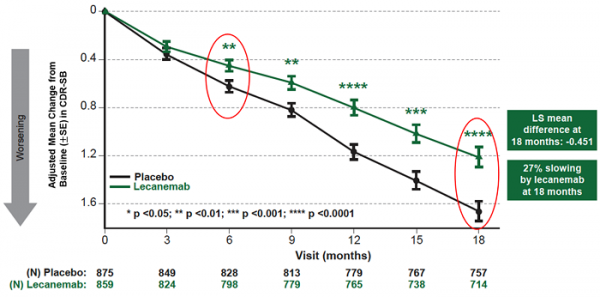
图1:主要终点cdr-sb变化值(18个月)
clarity ad安全性结果
仑卡奈单抗治疗组最常见的不良事件(>10%)是注射部位反应(仑卡奈单抗:26.4%;安慰剂:7.4%)、aria-h(合并脑微出血、大量脑出血和表面铁沉积症; 仑卡奈单抗:17.3%;安慰剂:9.0%)、aria-e(水肿/渗出性改变; 仑卡奈单抗:12.6%;安慰剂:1.7%)、头疼(仑卡奈单抗:11.1%;安慰剂:8.1%)以及跌倒(仑卡奈单抗:10.4%;安慰剂:9.6%)。注射部位反应绝大部分为轻中度(1~2级:96%),且主要发生于首次给药时(75%)。
在18个月的双盲试验期间,仑卡奈单抗组和安慰剂的死亡率分别为0.7%和0.8%,没有发现与仑卡奈单抗治疗或淀粉样蛋白相关的影像学异常(aria)相关的死亡。分别有14%和11.3%的仑卡奈单抗治疗组与安慰剂组的患者发现严重不良事件。治疗组与安慰剂组治疗期间报告的不良事件(teae)发生率分别为88.9%和81.9%,teae所致治疗组与安慰剂组的停药率分别为6.9%和2.9%。
总体上,根据 ii 期试验结果,仑卡奈单抗的aria发病率符合预期范围。在影像学上aria-e事件主要表现为轻中度 (占aria-e患者的91%)或无症状(占aria-e患者的78%),且主要见于治疗前3个月内(占aria-e患者的71%),并在检测出aria-e后4个月内自行缓解(占aria-e患者的81%)。2.8%接受仑卡奈单抗治疗的有症状aria-e患者中,最常见头痛、视觉障碍和意识错乱。在仑卡奈单抗组和安慰剂组中,有症状aria-h的发生率分别为0.7%和0.2%。在仑卡奈单抗(8.9%)和安慰剂(7.8%)之间,未观察到单独aria-h (即在未发生aria-e的参与者中发生aria-h)失衡。与apoe4携带者相比,aria-e和aria-h在apoe4非携带者中更少见,而apoe4纯合子携带者较apoe4杂合子携带者发生频率更高。在核心研究和随后的开放标签扩展研究中,安慰剂组(1/897)和仑卡奈单抗组(2/1608)合并脑出血的死亡率均为0.1%,其中接受仑卡奈单抗治疗的2例患者死亡发生在开放标签扩展研究中。2例患者均存在明显的合并症以及抗凝治疗导致的大出血或死亡的危险因素。因此,研究者评估认为该死亡病例与仑卡奈单抗治疗无关。
clarity ad研究影像学、血清和csf生物标志物评估几结果
评估了仑卡奈单抗治疗后的淀粉样蛋白、tau蛋白和神经变性的影像学、血清和csf生物标志物。仑卡奈单抗治疗后,csf和血清淀粉样蛋白生物标志物 aβ 42/40 比率提示早期和持续的逆转淀粉样蛋白的作用。仑卡奈单抗治疗18个月后,平均淀粉样蛋白pet为22.99 centiloids,低于淀粉样蛋白的阳性阈值(30 centiloids)。tau生物标志物评估提示,清除淀粉样蛋白可改善csf和血清p-tau (p-tau181)水平,而p-tau是ad病理通路中淀粉样蛋白的下游生物标志物。tau蛋白pet分析表明,与安慰剂相比,仑卡奈单抗治疗延缓了颞叶内的tau蛋白沉积,并改善了总tau蛋白(t-tau)沉积。根据神经变性的生物标志物评估,仑卡奈单抗改善了血清胶质纤维酸性蛋白(gfap,星形胶质细胞活化的标志物)和csf神经颗粒素(突触功能障碍的标志物),且经治疗后均恢复至正常水平,而仑卡奈单抗和安慰剂之间的csf或血清神经纤维轻链无显著差异。
clarity ad 结果
阿尔茨海默病是一种神经系统退行性疾病,严重影响患者及其家属。随着全球人口老龄化的加剧,ad已成为社会和医疗系统的一个重大问题,急需作用于疾病病理生理机制的新型治疗药物。早期ad的治疗目标是对认知功能、日常生活活动和精神症状产生持续影响,通过减缓疾病进展使患者维持更久的独立性,改善或维持生活质量。
在卫材的验证性ⅲ期研究clarity ad中,仑卡奈单抗在不同认知和功能量表以及亚组(人种、民族、合并症)之间证实了结果的一致性。根据cdr评估,仑卡奈单抗治疗使疾病进展位下一阶段的风险降低了31%(风险比:0.69)。基于观察到的数据和外推至30个月的cdr-sb进行的斜率分析表明,仑卡奈单抗治疗25.5个月相当于安慰剂18个月时的水平,这表明仑卡奈单抗可以延缓疾病进展达7.5个月。基于ⅱ期试验数据的模拟模型提示,仑卡奈单抗可能将疾病进展速度减缓2.5 ~ 3.1年,帮助患者可以更久的维持在ad的早期阶段。此外,仑卡奈单抗还能维持患者健康相关的生活质量,减轻照料者的负担(减少评分恶化23-56%)。认知和功能、疾病进展、健康相关的生活质量以及照料者负担等方面的证据共同表明,仑卡奈单抗治疗可为患者、照料者、临床医生和社会带来有积极的益处。
卫材正在网络直播本次会议,介绍仑卡奈单抗的情况,可以在卫材株式会社网站的投资者栏目中观看直播,之后还可以点播这些内容。
仑卡奈单抗的全球开发和监管提交由卫材主导,而产品则由卫材和渤健共同商业化和推广,其中卫材拥有最终决策权。
本新闻稿讨论的是一种正在开发的制剂的研究用途,并不打算传达关于疗效或安全性的结论。不能保证这种研究性药物将成功获得卫生部门的批准。
1 cdr-sb是一种用于量化痴呆症状的各种严重程度的数字量表。cdr-sb评分从患者记忆、方位、判断和解决问题的能力、家庭以外的活动、家庭和嗜好以及个人照顾六方面进行综合性评估。6个分项的总分即cdr-sb得分,cdr-sb也可作为评价早期ad治疗药物有效性的评估量表。
2 adas-cog是全球ad临床试验中最常用的认知评估工具。adascog14由14项能力组成:单词回忆、指令、结构式使用、物体和手指命名、概念使用、定向、单词识别、记忆单词识别指令、口语理解、找词困难、口语能力、延迟单词回忆、数字划消和迷宫任务。adas-cog 可以用于早期ad的评估,包括mci阶段。
3 adcoms由卫材开发,结合了adas-cog量表的认知功能评估、mmse和cdr量表的痴呆严重程度评估项目,能够高度敏感地检测早期ad临床功能的变化和记忆的变化。
4 adcs mci- adl基于对患者伴侣的24个关于近期实际日常生活活动的问题,用来评估mci患者的日常生活活动能力。